
Student-facing materials
Laura Corns; Andrew Metcalfe; and Elena Rainero
Introduction
Cell migration is crucial for many physiological processes including development, immune responses and wound healing. However, cell migration is also a key process in the pathophysiology of cancer. In this project you will investigate the migration of epithelial ovarian cancer cells using an in-vitro cell culture scratch assay. Specifically, you will be designing experiments to elucidate the role of different voltage-gated sodium channels in this process. This will enhance your understanding of the pathophysiology of cancer and will demonstrate the importance of cell biology in biomedical science.
It is important for you to be aware that this is a real scientific investigation; there are very few research articles that have investigated the role of voltage-gated sodium channels in ovarian cancer cell migration. The positive of this is that you could contribute experimental evidence to an unanswered question (super exciting!). The negative is that we do not know exactly what will happen when you apply different concentrations of different drugs to the cells, so you may see no effect; however, it is important to remember that ‘no effect’ can still be an important result. Finally, there is a strong emphasis on experimental design in this project, with you making decisions about the experimental hypothesis and protocol.
Project outline
Aims
- To design an appropriate experiment to investigate whether voltage-gated sodium channels influence the rate of cell migration in human epithelial ovarian cancer cells using an in-vitro cell culture scratch assay.
- To communicate your experimental design and findings via a scientific poster.
Learning Objectives
Following the successful completion of the project, you will be better able to:
- Develop an appropriate hypothesis based on the current scientific literature
- Design an experiment that effectively tests a hypothesis
- Identify and use appropriate negative and positive controls
- Evaluate the appropriateness of an experimental protocol and make modifications to a protocol based on preliminary results
- Use aseptic technique, a pipettor and an inverted microscope
- Calculate and perform the dilutions of drugs
- Choose and perform appropriate imaging and data handling techniques
- Evaluate results appropriately and put them into context
Who am I working with and what is the timeline of the project?
- This project will be performed within your tutor groups, in groups of 4. You can decide how the tutor group is split yourselves.
- As a group you will:
- be given two 6 well plates in session 1 and session 3.
- perform the preliminary experiments (sessions 1 and 2).
- formulate a hypothesis about the role of one or more voltage-gated sodium channels in cell migration of human ovarian cancer cells (in between session 2 and 3).
- select one or more drugs to test, and which concentrations you should use the drugs at (this will be limited to some extent by health and safety). This should be based on your hypothesis.
- perform your experiments (sessions 3 and 4).
Why do we have 2 sessions in one day?
- Session 1 – Set up preliminary scratch assay experiments, take initial images.
- Session 2 – Return later that day to take the second set of images, perform initial analysis and begin designing the next set of experiments.
- The sessions must be on the same day otherwise, too much cell migration may occur and you will not see a significant difference between control and experimental wells. The same is true for sessions 3 and 4 as the same process is repeated.
Background information
Before starting this section you should have attended the introductory lecture about cancer.
Cell migration
Now you know why cell migration is a critical part of cancer progression, we will review the key structures and processes that are essential for cell migration. Cell migration consists of three phases: protrusion, attachment and traction.
- Protrusion. Protrusion relies on the plasma membrane being pushed out due to the polymerisation of actin filaments which stretches the actin cortex of a cell. These protrusive structures can be known as filopodia and lamellipodia depending on their structure. Actin molecules are added to the positive end of the actin filament and removed from the negative end leading to a unidirectional treadmilling process which is regulated by a number of molecules.
- Attachment. The protrusions must then adhere to the substratum (extracellular matrix); this is achieved through integrin mediated adhesions.
- Traction. The rear of the cell must then be able to de-adhere from the substratum so that the rear of the cell can be pulled forward by traction with help from the stress fibres.
Many steps within this process are regulated by a group of monomeric GTPases that are members of the Rho protein family. This includes Rac, whose activation promotes actin polymerisation.
The in-vitro scratch assay
A quick and simple method of assessing cell migration is the in-vitro scratch assay. This involves making a scratch in a layer of cells and observing how well the gap closes between the two edges of the scratch. The faster and more completely the gap closes, the more efficient the cell migration has been.
This method does have its issues as it doesn’t replicate the complex extracellular matrix that contains various chemical and mechanical cues which influence migration. However, it is useful as an initial, simplified measurement of cell migration.
For further information on the scratch assay you may wish to consult this paper; Liang, C., Park, A.Y., Guan, J (2007). In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature Protocols, 2, 329–333.
Voltage-gated sodium channels
Voltage-gated sodium channels (VGSCs) are transmembrane proteins which are commonly associated with excitable tissues and are well known to be essential for action potential generation in neurons. However, VGSCs are also expressed outside of neurons, including in cancerous cells.
There are 9 alpha subunits NaV1.1 to NaV1.9, each encoded by a different gene. VGSCs are broadly referred to by which alpha subunit they contain. Each VGSC has different:
- expression patterns – i.e. is expressed in a different combination of cells around the body
- electrophysiological properties – e.g. some may have faster kinetics for opening than others
In general:
- NaV1.1, 1.2, 1.3, 1.5 and 1.6 are widely distributed in the CNS
- NaV1.5 is also expressed in cardiac tissue
- NaV1.7, 1.8 and 1.9 are distributed in the PNS
There are 4 beta subunits. These subunits can modulate the properties of the alpha subunit and can affect the plasma membrane expression of VGSCs.
For health and safety reasons, we have limited the type of voltage-gated sodium channel blockers available to you to those that are selective for subunits that are primarily expressed in the peripheral nervous system, avoiding NaV1.5 inhibitors due to their potential cardiac side effects.
The paper below has looked at the expression levels of different NaV subunits in some ovarian cancer cell lines which may inform your hypothesis regarding which VGSCs to investigate in sessions 3 and 4. However, note that none of the cell lines used in the paper are the one we are using so there may be differences. You do not need to look at this paper until you develop your hypothesis at the end of session 2.
Gao et al. (2010). Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncology reports, 23, 1293-1299.
What cells are we using?
You will be provided with two 6 well plates per group of 4. These plates will contain confluent monolayers of A2780-Rab25 human ovarian cancer cells. These cells were initially isolated from a patient with untreated ovarian endometrioid adenocarcinoma. They have subsequently been engineered to overexpress Rab25. Rab25 is a small GTPase that is involved in cell surface receptor recycling and the activation of cell signalling pathways. You do not need to factor Rab25 expression into your experimental design (it is being used in research labs within the department which is why we have this particular version of the A2780 cell line!).
Although you will not be culturing and plating the cells yourself, it is important that you understand this process and the importance of aseptic technique within it. Although you will not be able to apply all of the principles of the aseptic technique during this practical, you should still follow as many principles as possible to reduce the chances of your cells becoming infected.
Cell culture
Some cell lines require the bottoms of the wells to be coated in an extracellular matrix such as collagen to help them adhere (stick) to the well and grow. However, A2780-Rab25 are adherent cells, which means that they stick directly onto the bottom of the well without the need for an added matrix.
Passaging cells in culture
Once cells reach a certain confluency they can start to become unhealthy, therefore, they have to be resuspended in media, counted and plated at lower densities; this process is known as passaging and has been completed by our teaching technical team in order to plate your cells in their wells.
- To passage, cells should be detached from the surface of the culture dish using Trypsin-EDTA.
- Media containing serum acts as a Trypsin inhibitor. So, cells should be washed twice in phosphate-buffered saline (PBS) before adding Trypsin.
- Cells are then incubated at 37°C for 3 minutes to allow Trypsin to work.
- Trypsin is then neutralised by suspending the cells within the media containing serum. The media should be gently pipetted whilst rocking the dish to ensure the cells are dispersed equally.
- Determine the cell counts within the cell suspension by taking an aliquot and using the hemocytometer.
- Plate the required cells onto the 6-well plates with 2 ml of growth media per well. Incubating the plates at 37°C enables cells to attach and spread creating a confluent monolayer of cells.
Aseptic technique
Why do we need aseptic technique when handling cell cultures?
To prevent cell cultures from becoming contaminated by a variety of microorganisms, including bacteria, fungi and viruses, we must keep them in a sterile environment.
What are the sources of biological contamination?
- Nonsterile supplies e.g. petri dishes or plates, media and reagents
- Bacteria and fungi on hands and clothes
- Airborne particles which contain microorganisms
- Unclean incubators
- Dirty work surfaces.
What does aseptic technique do?
It provides a barrier between the potentially contaminating microorganisms and the sterile cell culture. This barrier reduces the probability that the cell culture will become contaminated.
What are the main elements of aseptic technique?
- A sterile work area
- Good personal hygiene
- Sterile reagents and media
- Sterile handling.
How will we be using aseptic technique?
Due to the limited availability of cell culture hoods you will be performing the experiment on the bench. As we will only be using the cells on the same day that they have been exposed to an unsterile environment, this will not be enough time for any contamination to substantially affect our experiments. Regardless, we will still aim to maintain as sterile an environment as possible.
You will be asked to maintain the following aseptic technique:
- A sterile work area:
- Clean your work areas with 70% ethanol
- Do not have any personal items on the bench
- Set your area up in a way that mimics the set up in the cell culture hood (minimise crossover for example).
- Good personal hygiene:
- Wash your hands
- Wear gloves and a clean lab coat
- Tie long hair back
- Sterile media:
- The media you are given will be sterile at the start of the class
- Wipe any bottles of media and eppendorfs with 70% ethanol
- Keep bottles and eppendorfs lids closed when not in use.
- Sterile handling:
- Work slowly and deliberately
- When opening bottles, keep the lid in your hand or place inside facing down
- Be careful not to touch the pipette to the outside of any bottles.
Experiment 1: sessions 1 and 2
Aim
This is your preliminary experiment which should be used to determine the most reliable and effective experimental design for your experiment in session 3.
Equipment
Each group of 4 should have a bench with the following:
- 2 x 6 well plates with confluent monolayers of A2780-Rab25 cells
- Fine marker pen
- p10 pipette and tips
- p200 pipette and tips
- p1000 pipette and tips
- pipettor (also known as a PIPETBOY)
- plastic serological pipettes (which are used with the pipettor)
- sterile media
- 1 x eppendorf of DMSO (10%). All of the VGSC inhibitor stock solutions available in session 3 will be dissolved in DMSO. This is a solvent that is known to directly effect some cell effects. Following dilution of the VGSC inhibitors from stock to working concentrations, the maximum concentration of DMSO that cells would be exposed to is 0.1% DMSO.
- 1 x eppendorf of 1mM NSC23766. NSC23766 inhibits Rac1 at a concentration of 25μM. Rac1 is a small GTPase that is essential for cell migration.
Experimental Protocol
As you are designing your own experiments, this protocol contains multiple options at each stage. Read each option and discuss as a group which one you wish to use and/or how you will test the various options. Some stages have minimum requirements that all groups should follow to ensure some results are collected. More in depth protocols about how to use the equipment can be found in the ‘Technical skills required for your experiments’ section.
Labelling plates
- Minimum requirement. Clearly label each plate so that you can identify it when you return in session 2. We suggest adding 2 sets of initials per plate.
- Ensure each well is also clearly labelled so you remember exactly what condition was tested in each well. This could be directly on the plate or using a numbering system on the plate and subsequent notes. Your lid should only fit in one direction, if this isn’t the case ensure that both the lid and bottom are labelled, otherwise you will mix up your wells.
Creating a scratch
- Minimum requirement. You have 2 x 6 well plates for each group of 4; this means that you have 12 wells to use in your preliminary experiments. Ensure each well has a scratch, don’t waste opportunities to trial different conditions or increase the number of repeats per condition.
- You can choose the same person to make every scratch or divide the wells equally between you. If dividing the wells, consider carefully how you will divide them.
- Scratches are made by dragging a pipette tip along the bottom of each well.
Choosing your controls
- Determine which wells will have negative controls – these are the controls which you do not want to have an effect.
- Determine which wells will have positive controls – these are the controls which you want to have an effect
Imaging your scratch
- Minimum requirement. Each scratch must be imaged in at least one location at both 0 hrs (as soon after all scratches are complete as you can use the microscope) and 4 hrs.
- Minimum requirement. We will be saving all images to a memory stick on the microscope and adding them to a shared folder on the Google Drive. Each image should be appropriately labelled so that you can identify it. We suggest:
- Tutors surname_Your surname_plate number_well number_timepoint
- If using the cross scratch approach, you could image more than one arm of the cross and the centre.
- If using the vertical scratch approach, you could take an image above the horizontal pen line and below.
Measuring your scratch
- Minimum requirement. Each scratch must have at least one measurement at each time point.
- Minimum requirement. You should attempt to use FIJI (ImageJ) to make your measurements.
- Minimum requirement. Scratches at 0 hrs and at 4 hrs should be measured in the same place. Your experimental design up to this point should enable this.
- You may choose to take more measurements of each scratch and make a mean +/-SD
Technical skills required for your experiments
To allow you to design your preliminary experiments, it’s important that you can see how certain aspects of the scratch assay are performed.
Labelling plates
- Minimum requirement. Clearly label each plate so that you can identify it when you return in session 2. We suggest adding 2 sets of initials per plate.
- Ensure each well is also clearly labelled so you remember exactly what condition was tested in each well. This could be directly on the plate or using a numbering system on the plate and subsequent notes. Your lid should only fit in one direction, if this isn’t the case ensure that both the lid and bottom are labelled, otherwise you will mix up your wells.
- Depending on whether you create a single vertical scratch or a cross using a vertical and horizontal scratch, you need to consider whether you want to add lines in pen to the underside of your plate. Do not wait until you have made your scratches and added the media/drugs to draw these lines. In previous years, if only performing a single scratch we have found the pen lines in the figure below to be useful. The key thing here is that you can accurately image the same part of the scratch at 0 hrs and 4 hrs.
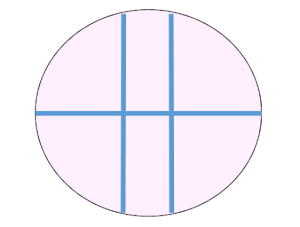
Example of lines drawn on the underside of each well. The scratch itself would be performed between the two vertical lines.
How to use a pipettor
You have all used a pipette before, but the majority of you will not have used an electronic pipettor. These are used extensively in cell culture and it is a useful skill to learn. It is worth practising moving media out of and into the container of media that you have been given to get a feel for how these work. Before moving media into your 6 well plate, you should practise your pipetting using the following instructions.
- Partially unwrap the serological pipettes (those very long ones!) but do not touch the plastic pipette with your hand. Once the top is open, hold the pipette on the outside of the wrapper and push into the pipettor. As we are not too concerned about sterility today, do not worry if you end up touching the plastic of the actual pipette, however, in an experiment where sterility was key, you would have now compromised the sterility.
- Place the pipette into the media and use the buttons on the handle to automatically move the media. Be extra careful not to fill the whole pipette and get the filter at the top of the pipette wet; this would compromise sterility and accuracy of the pipettor.
- If the filter gets wet i.e. it turns pink, replace immediately. Be careful for bubbles, taking a mix of air and media in at the start will move bubbles up the pipette and wet the filter.
Creating the scratches
- Minimum requirement. You have 2 x 6 well plates for each group of 4; this means that you have 12 wells to use in your preliminary experiments. Ensure each well has a scratch, don’t waste opportunities to trial different conditions or increase the number of repeats per condition.
- You create a scratch in your wells by scraping a pipette tip along the monolayer of cells. You should apply firm pressure but not so hard that you create a groove in the underlying plastic of the well.
- You choose what size pipette tips.
- You choose whether to do a single vertical scratch OR to create a cross with a vertical scratch and a horizontal scratch.
- You choose whether everyone has a go or you stick to the same person.
- Using the pipettor (see section above) remove the media from one of your wells and discard in your waste bucket. Important: do not leave your cells without any media for very long (seconds is OK, minutes is not), this is not healthy for them.
- Use the pipettor to add ~1 ml of media and swirl the media around the wells and then remove and discard this media.
- As the scratch disturbs some cells and causes them to detach, it is important that these cells are removed by removing the original media. The additional 1 ml helps to wash away any remaining detached cells.
- You can repeat this step if you want to do a double wash.
- Use the pipettor to add exactly 2 ml of control media i.e. the media you have been using so far.
- If you feel confident using the pipettor, you can do all of the wells in one plate at once (i.e. do step 2 for all wells, then step 3 for all wells, rather than steps 1-3 for one well, then steps 1-3 for a second well), however, only do this if the cells will not be left without media for less than a minute.
Adding your solutions
- You must now add your chosen solutions to each well.
- Note that each well already has 2 ml of control media and you may wish to simply leave some wells like this.
- For wells in which you are adding another negative control, a positive control or in session 3 an experimental drug, you will be adding the appropriate volume of stock solution directly to the well.
- To ensure your calculations are accurate, remember that your final volume should be 2ml.
- Once you have added the appropriate volume of stock solution to the well, give the plate a gentle swirl to mix the drug.
- Once all 6 wells have scratches and the appropriate solutions added, it is time to image your scratches. Although we would like the scratches to be imaged at 0 hrs (i.e. as soon as a microscope is available after creating your scratches).
Imaging your scratch
- Minimum requirement. Each scratch must be imaged at at least one location at both 0 hrs and 4 hrs. Note the time at which you take your initial images.
- If using the cross scratch approach, you could image more than one arm of the cross and the centre.
- If using the vertical scratch approach, you could take an image above the horizontal pen line and below.
- The microscope you will be using is a digital, inverted microscope; the particular ones that we are using are called EVOS.
- To take your image you need to:
- Place your plate on the stage
- Select the transmission light
- Set the objective at the lowest magnification to start with
- Use the controls to move the stage and adjust the focus to find your cells
- Once you have found your cells and identified your scratch, adjust the objective until you are happy with the image
- Capture your image
- Save your image with an appropriate name. Tutors surname_Your surname_plate number_well number_timepoint
Designing your preliminary experiment
Discuss how you will perform your preliminary experiment in your group of 4 and complete the experimental plan for today’s session below. Please ensure that it is checked by a member of staff (academic or GTA) before you start your scratches.
For each well consider:
- What size scratch is most appropriate?
- Scratches can be made with P10, P200 and P1000 pipette tips.
- How can I make sure that I am imaging the same place at both time points i.e. in session 1 and session 2?
- Some people create a cross with a vertical scratch and horizontal scratch, imaging each ‘arm’ of the scratch plus the centre.
- Other people add horizontal (and sometimes also vertical) marker pen lines on the underside of the well and then create one vertical scratch, taking images with the pen line in the field of view.
- What solutions are you using?
- Media alone, DMSO or NSC23766.
- Controls are an essential part of experiments.
- Negative controls test that any other experimental variables are not influencing the outcome of our experiment e.g. if a solvent such as DMSO is used to dissolve our drugs, then we need to check this solvent does not influence cell survival and migration.
- Positive controls show that an effect is possible i.e. a known inhibitor or enhancer of cell migration would be useful to verify that cell migration can indeed be inhibited or enhanced in these cells.
- How do you know whether a negative or positive control has worked?
Use this table to plan which solutions will be in which wells in your 2 x 6 well plates.
| Column 1 | Column 2 | Column 3 | |
| Plate 1 Row 1 | |||
| Plate 1 Row 2 | |||
| Plate 2 Row 1 | |||
| Plate 2 Row 2 |
Use this space to write your protocol considering the order in which you are going to perform the different steps
Solution Concentrations
You may find the table below useful to calculate any required volumes of drugs required. You should remember to consider the units and orders of magnitude that you are working in and how you write them in the table. You do not need to use all 3 rows.
Consider whether you will need to remove any media from the 2ml already in the well to ensure that the final volume once the drug has been added will still be 2 ml.
| Stock concentration | Working concentration | Stock volume | Final volume | |
| Solution 1 | ||||
| Solution 2 |
Session 2: Imaging scratches at 4 hrs and measuring cell migration
Choosing which voltage-gated sodium channel modulators to investigate
Now that you have started your preliminary experiments, in your groups, you can start to develop your hypothesis surrounding the role of voltage-gated sodium channels in cell migration of ovarian cancer cells. Depending on the time it has taken you to complete the first step of your preliminary experiments, you can either start the research into the literature in either lab 1 or lab 2. You do not need to make a decision today. Be targeted in your reading, you don’t need to read everything, select what is relevant to the drugs that are available to you.
Here is the review paper mentioned in the introductory lecture: Mao et al., (2019). The emerging role of voltage-gated sodium channels in tumor biology. Frontiers in oncology, 9(124).
Here is a reminder a paper which has investigated VGSC expression levels in ovarian cancer cell lines: Gao et al., (2010). Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncology reports, 23, 1293-1299.
Below is a table of the potential drugs that you can use, the maximum values are there for safety reasons. As long as you wear the appropriate PPE and apply due care whilst using the drugs, you will be perfectly safe. You can use these drugs alone or together.
| Drug | Target | IC50 for target | Suggested concentration | Selectivity | Safety |
| PF-05089771 | Nav1.7 inhibitor | 11 nM | Suggested 50 nM
Max 1µM |
IC50 values are 0.11 μM for Nav1.2, 0.16 μM for Nav1.6, 0.85 μM for Nav1.1, 10 μM for Nav1.4, 11μM for Nav1.3 and 25 μM for Nav1.5. | Possible irritation of throat, skin and eyes if contact made |
| PF-04531083 | Nav1.8 inhibitor | Data unpublished. | Suggested 1 µM | Data unpublished. | H302 Harmful if swallowed.
Currently in clinical trials for pain relief at low doses. |
| Epidermal growth factor (EGF) | Can upregulate Nav expression | N/A | 30ng/ml | Unknown | Non |
Imaging your scratches at 4 hrs
- One person from each group should notify a member of staff 10 minutes before the 4hr time point so that they can take you to collect your plates from the incubator.
- Once collected you should bring your plate to one of the microscopes. You do not need to use the same microscope as before but you may find it easier to do so. However, you must take images with the same objective so that the magnification is the same and they are easily comparable to the 0 hr ones.
- At the microscope you should aim to take an image of the same area of each scratch. This should be identifiable due to the use of pen lines or the cross shaped scratch.
- Save this image. You need to be able to distinguish this image from the image you took earlier today:
- Tutors surname_Your surname_plate number_well number_timepoint
Using ImageJ (FIJI) to measure your scratches
- Open the ImageJ (FIJI) software. This should already be downloaded however, if it isn’t you can download it here Fiji Downloads (imagej.net) . Once the folder has downloaded, click extract all and then double-click the microscope icon called ImageJ-win64.
- Drag and drop your first image into the software. Alternatively, click file, open and select your image from where you saved it (probably in downloads or desktop).
- If you’d like to adjust the brightness/contrast of the image, you can click on Image, go down to Adjust then across to brightness/contrast. This will bring up a new window where you can adjust the slide bars to change the settings of the image. Take care not to adjust the settings too much, it shouldn’t look artificial but just a clearer version of the original image.
- Set the scale so that you are measuring in micrometers not pixels.
- Although some microscopes add the scale bar directly to your images, these are not always calibrated, therefore we would recommend that you use the images of the stage micrometers provided (see figure below) to set the scale.
- On the stage micrometers that have been imaged, each division is 100 µm, therefore the distance between 0 to 10 (see figure below) is 1000 µm.
- Open the image of the stage micrometer in addition to your scratch image.
- Use the Line Selection tool to draw a line between a known distance e.g. 0-10 on the image of the stage micrometer.
- Go to menu – Analyze – Set Scale
- Set the known distance of the line you have drawn in the “Know Distance” Field. The length of the line in pixels will automatically be recognised in the tool.
- Set the units of the measurement. We would suggest µm.
- Check ‘Global’. This ensures that all open images will use the same spatial calibration once you click OK.
- You can check which images have been calibrated by looking at the information bar at the top of each image. If you draw a line on an image and pixels show up in the information bar rather than µm then it has not been calibrated.
- Once you are happy with the image, click on an appropriate tool and measure the distance of the scratch wound. Click Measure or Ctrl+M after each measurement to bring up a table with the measured values in it. You only need to record the value for length. Once all the measurements are complete, you can select all and copy. Open excel and paste the table into a blank spreadsheet.
- Repeat this for your second image and paste the values into the same spreadsheet making sure you label 0 hrs and 4 hrs separately. You can now perform any descriptive statistics you require in excel or import into R for data analysis.
Planning the next experiment
Now it’s time to consider what these results mean for your next experiment. Consider:
- Size:
- Which size scratch produced the more useful results?
- Did any heal too quickly?
- Any too large to fit on the microscope screen?
- Pattern:
- Were you happy with the type of scratch and your method for ensuring you could identify the same place?
- What pattern of scratch will you use in your next experiment?
- Will you use any pen markings?
- Controls:
- What happened in the negative control wells? Did these go to plan?
- What negative controls will you do in your next experiments?
- What happened in the positive control wells? Did these go to plan?
- What positive controls will you do in your next experiments?
Developing your hypothesis and experimental protocol
Now that you have had a chance to trial some different conditions for the scratch assay you should be able to refine your protocol for your next experiment. This should be combined with your research of the VGSCs to develop a hypothesis which you can test.
- What is your hypothesis with regards to VGSCs and cell migration in the ovarian cancer cell line A2780-Rab25?
- What is your protocol to test this hypothesis?
Experiment 2: sessions 3 and 4
Aim
- Perform your experiments to test the hypothesis that you have developed regarding the role of voltage-gated sodium channels in the migration of A2780 cells.
- Start to develop your poster.
Your experiments
- Before you start your experiments today, please ensure that you have checked your protocol, including dilutions, with a member of staff.
- You have all of the same equipment that you had in Lab 1 available to you with the addition of the voltage-gated sodium channel (VGSC) inhibitors and EGF.
- When you are ready, please come and collect your 2 x 6 well plates from the front.
Health & Safety
The VGSC inhibitors and EGF will be kept in small aliquots at the front of the class. Please ensure that you have read the relevant information in the COSHH form before using these drugs.
When you are ready to use the drug, please inform a member of staff and they will provide you with the drug.
Your analysis: analysing your raw data
We are not going to specify which software you should use to analyse your data; you can choose to use Microsoft Excel, R, GraphPad Prism or any other reliable software that you feel confident with.
We would like you to produce:
- descriptive statistics to describe your data.
- inferential statistics to determine whether any of your variables caused a significant effect on the amount of cell migration.
Please find the time in this session to discuss what type of statistics you will be using with a member of staff and what type of graph you will use to display your data.
Evaluating your data
- Are any of your results statistically significant?
- Can you accept your hypothesis?
- If your results are not significantly different, can you explain why? Do you think it is due to experimental design or due to the underlying science? What would you do differently in a future experiment?
- If your result was significant, what experiment would you perform next?
- How do your results fit within the context of the literature?
