
Student-facing materials
Janet Cronshaw; Elizabeth Alvey; Catherine Heath; Qaiser Sheikh; Melanie Stapleton; and Ilona Willson
1. Overview
Over the next three lab sessions you will use CRISPR/Cas9 gene editing technology to knock out a gene involved in methionine biosynthesis (MET15) and replace it with a gene that confers resistance to an antibiotic called nourseothricin (NatMX). Successful gene editing will result in a yeast that will be unable to grow in the absence of methionine and will be resistant to nourseothricin. You will screen yeast for these phenotypes to determine the success of gene editing. These experiments use plasmids developed in a School of Biosciences research lab (Daniels et al., 2018).
Aims and objectives
These practicals aim to:
- Demonstrate how CRISPR/Cas9 gene editing technology can be used to modify the genome of Saccharomyces cerevisiae.
- Provide training in designing single guide RNAs for gene editing.
- Demonstrate how online databases can be used when designing experiments.
- Provide training in primer design for DNA analysis.
By the end of these practical sessions, you will have carried out the following experimental techniques:
- Transformed DNA into S. cerevisiae.
- Screened yeast transformants for resistance to nourseothricin.
- Screened yeast transformants for methionine auxotrophy.
- Designed a single guide RNA (sgRNA).
- Designed PCR screening primers.
- Kept an accurate and thorough record of your work in a lab book.
2. Practical Class Schedule
There will be three practical sessions, summarised in the table below:
| Session | Tasks to carry out |
| Pre-lab preparation | Read the Background Information and Experimental Overview (in this document).
It would be a good idea to review [lecture material on CRISPR] before this session. Read through the protocols for each day before coming to the relevant session. |
| Day 1 | Prepare competent S. cerevisiae.
Transform S. cerevisiae with the CRISPR plasmid and the repair template. Plasmid annotation exercise to understand the function and features of the two plasmids. |
| Day 2 | Calculate yeast transformation efficiency.
Set up screens for methionine auxotrophy and nourseothricin resistance. Design a single guide RNA (sgRNA). |
| Day 3 | Analyse the nourseothricin resistance screen.
Analyse the methionine auxotrophy screen. Design PCR screening primers. |
3. Background Information
CRISPR/Cas9 in Bacteria
CRISPR/Cas9 gene editing technology is adapted from a bacterial system that evolved to protect bacteria from invading organisms, such as bacteriophage (Doudna & Charpentier, 2014). How this works in bacteria was covered by [relevant lectures]; here is a quick overview to help you understand the practical, but you will find it useful to look back at the relevant lecture.
The genomes of certain bacterial species harbour a DNA sequence known as a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR). The repeats in this region originated from the genome of an invading organism, such as a bacteriophage. Transcription of these CRISPR sequences results in short RNA molecules (CRISPR RNA, crRNA) that are complementary to the genome of the invading phage.
In addition to the CRISPR repeats, bacteria also encode an RNA-directed DNA endonuclease. The nuclease binds to the crRNA and is guided to the specific DNA sequence that is complementary to the crRNA. Once bound, it cleaves DNA to induce a Double Strand Break (DSB).
There are several of these nucleases from different species. The one used most in molecular biology applications is an enzyme called Cas9 from Streptococcus pyogenes.
CRISPR/Cas9 Gene Editing Technology
Molecular biologists have adapted the bacterial CRISPR/Cas9 system as a tool to target genes in a sequence-specific manner. The coding sequence of the Cas9 protein has been cloned into plasmids that can express Cas9 in a variety of organisms (in these practicals, we will express Cas9 in S. cerevisiae).
Molecular biologists also manipulate the sequence of the crRNA to target Cas9 to a desired DNA sequence. In molecular biology applications, RNA that binds Cas9 is known as a single guide RNA (sgRNA). The sgRNA incorporates an RNA sequence that is equivalent to the crRNA and binds to the DNA target sequence. The sgRNA also incorporates scaffolding RNA sequences that are required for binding to Cas9 (in bacteria, this is mediated by a separate RNA called the tracrRNA).
Expression of a suitable sgRNA (along with the Cas9 protein) in an organism should result in a DSB at a unique DNA sequence. DSBs are a particularly toxic form of DNA damage that often result in cell death. Cells have two main mechanisms to repair DSBs:
- Non-Homologous End Joining (NHEJ). Essentially, the two broken ends of DNA are stuck back together again (it is a little bit more complicated than that!) This pathway is prone to errors, particularly small insertions or deletions of bases, which can disrupt the reading frame, resulting in a non-functional gene.
- Homology-Directed Repair (HDR). The cell uses a homologous DNA molecule (for example, the other chromosome of a pair) as a template. This repair pathway is error-free and does not result in mutations.
Depending on which pathway is used, gene editing can result in different changes to genes, such as disruption or editing:
Gene Knockout. If a Cas9-induced DSB is repaired by NHEJ, it is likely that mutations (often small deletions and/or insertions) will be incorporated into the repaired DNA. This can render the gene non functional, for example by disrupting the reading frame, causing missense mutations in essential amino acids, or resulting in a premature stop codon being encoded. Thus, CRISPR/Cas9 is a valuable tool for knocking out genes in a variety of organisms.
Gene Editing. It is possible, however, to use CRISPR/Cas9 to make more specific changes in vivo, by providing the cell with a repair template for it to use in HDR. The repair template encodes the DNA sequence that you wish to replace the endogenous sequence with. This can be anything from a single nucleotide change up to the insertion of a large region of DNA.
It is important to note that, although we can quite accurately control where the DSB occurs (via the sgRNA), we can’t control which pathway is used to repair the DSB. We can attempt to influence it (for example, by providing / not providing a repair template) but different cells will use different pathways. As this will result in different outcomes in different cells, screening is always required to confirm the desired outcome.
References
Daniels, P. W., Mukherjee, A., Goldman, A. S. & Hu, B. (2018) A set of novel CRISPR-based integrative vectors for Saccharomyces cerevisiae. Wellcome Open Res, 3, 72.
Doudna, J. A. & Charpentier, E. (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 346(6213), 1258096.
4. Experimental Overview
Gene Editing Strategy
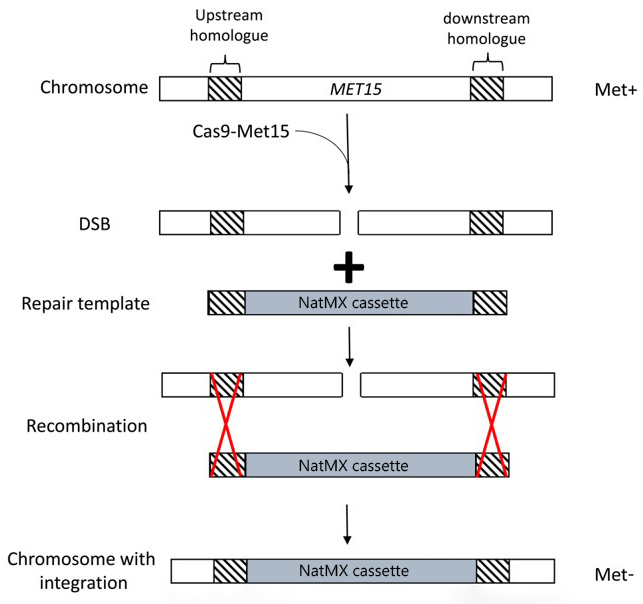
The overall strategy for these experiments is described briefly below and outlined in the figure.
- S. cerevisiae are transformed with two plasmids:
- A plasmid that expresses the nuclease Cas9 as well as an sgRNA that targets the MET15 gene.
- A plasmid containing our desired repair template (a gene conferring resistance to the antibiotic nourseothricin) flanked by regions homologous to the MET15 gene.
- The sgRNA-guided Cas9 enzyme induces a DSB in the MET15 gene.
- The repair template (the NatMX cassette) is used to repair this DSB, resulting in replacement of the MET15 gene by the NatMX cassette. The NatMX cassette confers resistance to an antibiotic called nourseothricin.
It should be noted that neither Cas9-mediated DNA cleavage nor HDR are 100% efficient! You will get many transformants and lots of them will not have undergone successful gene editing. Screening of transformants for the correct phenotype is vital.
Screening
S. cerevisiae transformants will be screened for successful replacement of the MET15 gene by the NatMX cassette in two ways:
- Methionine auxotrophy. The parental yeast strain can make its own methionine (referred to as Met+) and can therefore grow on media that does not contain methionine. Successful disruption of the MET15 gene would result in a yeast cell than can no longer make its own methionine (met−) and can only grow when methionine is added to the growth media.
- Nourseothricin resistance. The parental yeast strain is not resistant to the antibiotic nourseothricin (NAT−). A yeast cell that has successfully undergone gene replacement will be resistant to this antibiotic (NAT+).
In summary, the parental yeast strain (prior to gene editing) will have the phenotype Met+ NAT−. An edited yeast strain will be met− NAT+
Plasmids Used
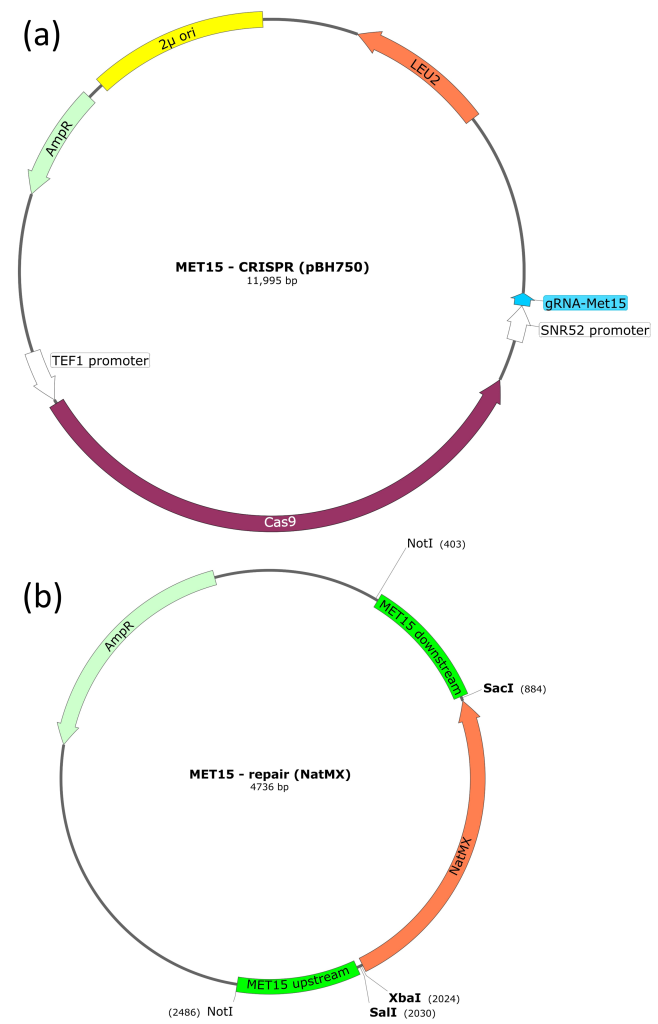
pBH750 (CRISPR plasmid)
This plasmid encodes the Cas9 protein, which cleaves DNA to produce a DSB. Cas9 is guided to a specific DNA sequence by an sgRNA that is also encoded by this plasmid. The chosen sgRNA is complementary to a region of the MET15 gene. Therefore, transformation of this plasmid into yeast cells should result in a double strand break (DSB) in the MET15 gene.
In addition to Cas9 and sgRNA coding regions, pBH750 also has an ampicillin-resistance gene (for selection and maintenance in E. coli) and a LEU2 gene. The LEU2 gene is also a selective marker but for S. cerevisiae (rather than E. coli). If we use a parental yeast strain that is leu2–, we can select for yeast that contain the pBH750 plasmid.
pBH763 (repair template)
This plasmid contains the NatMX cassette, which confers resistance to the antibiotic nourseothricin. Flanking the NatMX cassette are regions of homology to the MET15 gene, which mediate homology-directed repair (HDR). Flanking the MET15 homologous sequences are NotI restriction sites (these are important when preparing the plasmid for transformation into S. cerevisiae).
The plasmid also has an ampicillin-resistance gene (for selection and maintenance in E. coli).
5. Practical schedule
Day 1
Overview
- Prepare competent S. cerevisiae.
- Transform S. cerevisiae with the CRISPR plasmid (pBH750) and the repair template (pBH763). (The repair template needs to be linearised prior to transformation by using the restriction enzyme NotI. Owing to time constraints, we have already done this for you).
- Plate out to select for transformants. The S. cerevisiae strain that you are using today (K699) is leu2−. This means that the pBH750 plasmid can be selected for, since pBH750 carries the LEU2 gene as a selectable marker.
Lab protocols
Step 1: prepare competent yeast cells
You are provided with a 10 ml culture of yeast K699 cells (a leu2− strain) that have been growing overnight in YAPD. When the OD (at 600 nm) of the culture reached 0.4 – 2.0, the cultures were placed on ice to stop them growing. There are approximately 1×106 cells per ml for an OD600 of 0.1.
- Centrifuge the cultures for 10 minutes at approximately 4,500 rpm to pellet the cells. Carefully pour off the supernatant into a virkon pot.
- Resuspend the cell pellet in 1 ml of 0.1 M lithium acetate and transfer into a 1.5 ml Eppendorf tube.
- Centrifuge for 1 minute at 13,000 rpm and discard the supernatant into virkon.
- Wash the cells twice more in 1 ml 0.1 M lithium acetate (by repeated resuspending, centrifugation, and removal of the supernatant).
- Resuspend the final pellet in 0.4 ml 0.1 M lithium acetate.
- Incubate at 30°C for 30 minutes.
While your cells are incubating, prepare the DNA to be transformed (step 2).
Step 2: prepare the DNA to be transformed
Label three 1.5 ml Eppendorf tubes (1, 2 and 3 is fine but it’s up to you!) Add the following reagents to the three tubes:
| Transformation 1 | Transformation 2 | Transformation 3 |
| 5 μl (1 μg) pBH750
30 μl (1.5 μg) NotI linearised pBH763 15 μl water |
5 μl (1 μg) pBH750
45 μl water |
50 μl water |
- Once your competent cells are ready, add 50 µl of competent cells to each transformation tube.
- Then, to each transformation tube, add:
- 240 µl of 50% (w/v) Polyethylene Glycol (PEG)
- 35 µl of 1.0 M lithium acetate
- 25 µl of single stranded denatured salmon sperm DNA (10 mgml-1).
- Mix gently by pipetting up and down and incubate at 30°C for 20 minutes.
- Heat shock at 42°C for 20 minutes.
- Harvest cells by centrifugation in a microfuge (3 minutes at 6,000 rpm).
- Discard the supernatant and gently resuspend the cells in 1 ml YAPD, then transfer to sterile universal tubes (yellow lids).
- Incubate at 30°C for 60 minutes with gentle shaking (it might help to use an elastic band to group your tubes before placing in the shaking water bath).
- Transfer the culture to 1.5 ml tubes and harvest by centrifugation (30 seconds at 13,000 rpm) and discard the supernatant into virkon.
- Wash off the media by resuspending the cells in 1 ml sterile water. Centrifuge as above and discard the supernatant into virkon.
- Resuspend cells in 100 µl of sterile water.
- Spread the cells onto −leu plates and incubate at 30°C for 2-3 days. You will analyse transformation in the next session.
During the incubation steps
Find out about the MET15 gene
The sgRNA in the plasmid you are using today targets the MET15 gene. In a yeast cell that has been successfully edited, the MET15 gene will be non-functional; it might be useful to find out what that means! Go to the Saccharomyces Genome Database (SGD) (a database of information about S. cerevisiae) and search for MET15. Once you have found MET15, answer the following questions.
- MET15 has a few different names (genes often do if they were discovered by different people under different circumstances). What are some of the alternative names for MET15?
- What is the function of the protein encoded by the MET15 gene?
- What is the Name Description of the MET15 gene? What do you think this means about the phenotype of a yeast cell that is a met15 knockout?
Find out about the plasmids
You have a printout of the two plasmids used in these experiments.
- Research the features on the plasmids and annotate your printout to explain the role of each of them. To help you, you can use Google / consult Daniels et al., 2018 / ask a demonstrator.
- The repair template needs to be linearised prior to transformation into S. cerevisiae. Why do you think this might be?
Predict your results
Copy the table below into your lab book and fill in the table below to predict the phenotype you are expecting for each transformation, with respect to the three main genes. This can be difficult to get your head around as there are lots of minuses and pluses:
- Leu auxotrophy. A yeast cell that can grow on medium that does not contain leucine is wild-type (Leu+). A yeast cell that cannot grow on medium that lacks leucine is auxotrophic for leucine (leu–). It gets complicated with nomenclature because sometimes, when referring to the plates that lack leucine, we might call them –leu plates for short. This is not the same as the yeast phenotype (leu–) or the yeast genotype (leu2–).
- Met auxotrophy. Similarly, a yeast cell that can grow on medium that does not contain methionine is wild-type (Met+). A yeast cell that cannot grow on medium that lacks methionine is met–.
- Nourseothricin resistance. A yeast cell that can grow on medium containing nourseothricin is resistant to the antibiotic (NAT+). A yeast cell that cannot grow on medium containing nourseothricin is sensitive to the antibiotic (NAT-).
| Transformation | pBH750 | pBH763 | Leu phenotype | Met phenotype | NAT phenotype |
| T1 | yes | yes | |||
| T2 | yes | no | |||
| T3 | no | no |
Day 2
Overview
- Calculate yeast transformation efficiency.
- Set up a screen for methionine auxotrophy.
- Set up a screen for nourseothricin resistance.
- Design a single guide RNA (sgRNA).
Lab protocols
Calculate the Transformation Efficiency
Transformation efficiency is a measure of how well cells can take up DNA from their environment and express the genes encoded by that DNA. The units of transformation efficiency are Colony Forming Units (CFUs) per µg of DNA, and it is calculated as follows:
Transformation Efficiency = Number of Colonies / Mass of DNA (μg)
- Count the number of colonies on plates 1 and 2. If you have a lot of colonies, there is a tally counter in your drawer and you can use a marker to keep track of which colonies have been counted.
- Have a look at the information from day 1 to remind yourself of how much DNA you transformed. For transformation 1, you should calculate the transformation efficiency using the mass of pBH750 alone. This is because pBH750 is the plasmid that you selected for.
- Use the equation above to calculate the transformation efficiency for transformations 1 and 2 (transformation 3 had no DNA! If you have colonies on here, they are probably contamination).
Set up the phenotype screens
You should (hopefully!) have several colonies on your plate from transformation 1. If you have no colonies, don’t worry – it is fine to work with your neighbours and share plates, if they have colonies to spare. We also have rescue plates if there are a lot of people with no colonies.
In a perfect world, 100% of these colonies would have undergone gene replacement of the MET15 gene by the NatMX cassette. However, molecular biology is never 100% efficient and there are multiple possible outcomes (all of which will probably be represented on your plate). Screening of colonies for the appropriate phenotype(s) is always required. This is then usually followed by genotype screening, using a technique such as PCR.
The phenotypes you will screen for are methionine auxotrophy and nourseothricin resistance. These will allow you to identify colonies that have undergone successful gene replacement. In session 3, we will go through how to analyse the screening results in more detail and how to use the screening results to assay for gene editing. Use sterile technique when setting up your screens – you don’t want to contaminate your yeast colonies.
Methionine auxotrophy
This screen tests whether a colony can make its own methionine (phenotype = Met+) or whether it can only grow in the presence of an exogenous methionine supply (phenotype = met−).
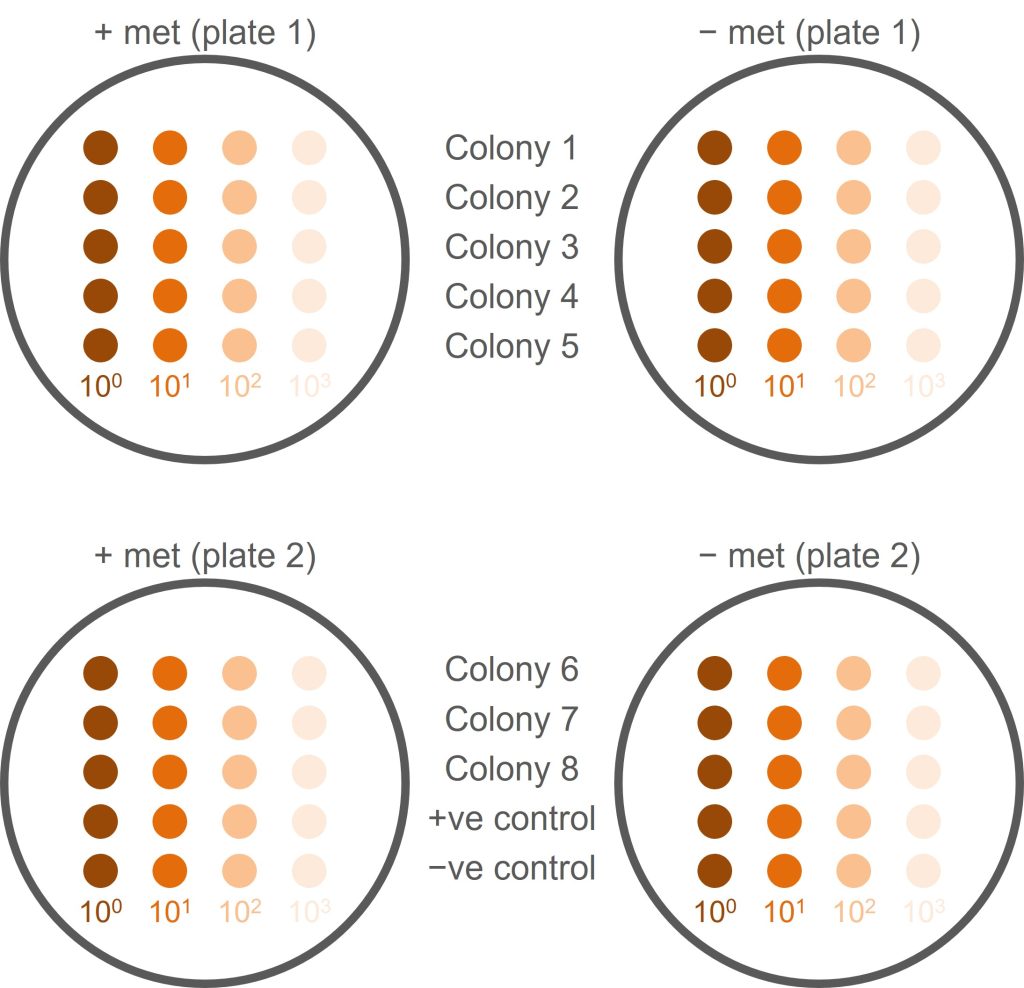
This screen is a little bit more complicated than just plating your colony onto a plate lacking methionine. This is because met– yeast cells will already contain some methionine which allows them to grow until the supply of methionine is used up. When the cells are at high density, this means that a phenotype is not visible, simply because there are a lot of cells. We therefore need to make a serial dilution of each yeast colony that we want to screen and compare the growth of serial dilutions.
An overview of how we will set up the screen can be seen in the figure below.
- Pick a healthy yeast colony (from transformation 1) and resuspend in 100 µl sterile water (do not use broth as this contains methionine, which might allow met− cells to grow). This is your undiluted (100) yeast suspension.
- Make a 10x dilution of your original yeast suspension by adding 10 µl of yeast suspension to 90 µl of distilled water. This is your 101 dilution.
- Make a 102 dilution by adding 10 µl of 101 dilution to 90 µl of water. Then make a 103 dilution by adding 10 µl of 102 dilution to 90 µl of water.
- For this single colony, you should now have four tubes: undiluted (100) yeast, 101, 102, and 103 dilutions. (It would now be a good idea to read ahead for the next few steps and plan accordingly!)
- You need to repeat the serial dilution for seven more colonies (screening eight colonies in total) and for a negative and a positive control. Controls are provided for you (negative control = parental K699 strain; positive control = K699 NatMX+).
- For each colony, spot 5 µl of each dilution onto a +met plate and 5 µl onto a −met plate. Use the figure below as a guide to how to organise this and use a numbered grid to help guide your pipetting and avoid cross-contamination.
- Do not discard the undiluted (100) cell suspension! You will need this for the nourseothricin resistance screen (next step).
- In total, you will have 10 colonies and four dilutions of each colony which is a lot of tubes! Coordinate this within your pair to make sure you keep track of what goes where and you don’t lose or mix up samples.
- Label the plates! You need to know whose plate it is, what type of plate it is, what is on the plate, where on the plate it is.
Nourseothricin resistance
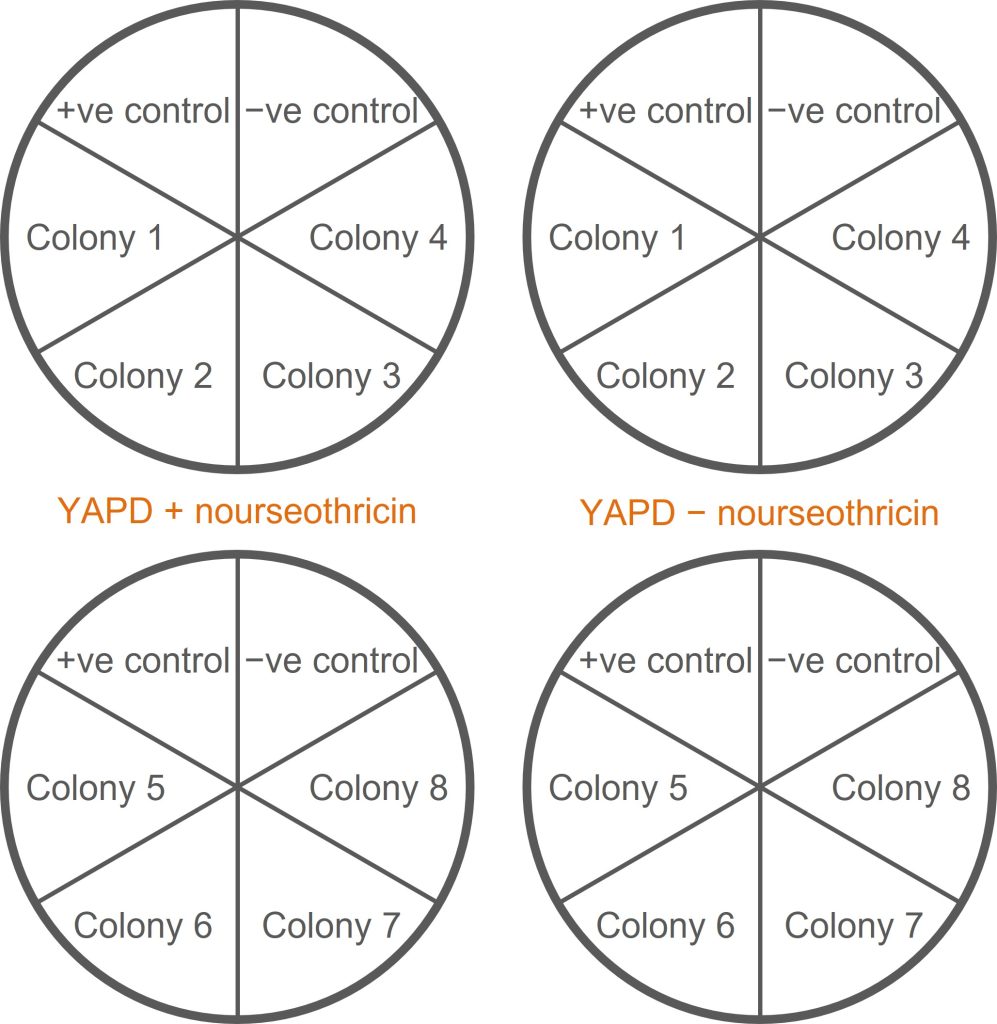
This screen tests whether a colony is resistant to the antibiotic nourseothricin (NAT+) or sensitive to nourseothricin (NAT−).
- You should have ten tubes remaining from the methionine auxotrophy screen: an undiluted yeast cell suspension from eight of your transformants; one from a negative control; and one from a positive control.
- Label two duplicate sets of YAPD plates: two containing nourseothricin (+nrs), two without nourseothricin (−nrs). Each plate should be divided up into six segments to accommodate six colonies (see the figure below for guidance on how to organise this).
- Use a wire loop to streak each cell suspension onto a segment of an +nrs plate. Repeat the streaking process (with the same cell suspension) onto an −nrs plate.
- Onto each plate, also streak the negative and positive controls (K699 and K699 NatMx+).
- Put these plates (and the plates from the methionine auxotrophy screen) into the bay trays for incubation at 30°C.
Design a single guide RNA (sgRNA) for MET15
You will be provided with a printout for this part of the session. [see Resources for Teachers section]
Day 3
Overview
- Analyse nourseothricin resistance and methionine auxotrophy screens from day 2.
- Design PCR screening primers.
- Finish off anything you didn’t get round to in previous sessions (plasmid annotation exercise; sgRNA design activity). This is your chance to ask questions about these things while we are still here to answer them!
Lab protocols
Before you analyse your colonies
Construct a table in your lab book to record your screen data (here is an example, but you are free to design your own table with whatever information you would find helpful):
| Colony | Met phenotype | NAT phenotype | Genotype | Gene editing outcome |
| Negative control (parental K699) | growth | no growth | Met+ NAT- | WT genome – no gene editing. |
| Positive control (K699 NatMX+) | no growth | growth | met- NAT+ | MET15 gene has been successfully replaced by the NatMX cassette. |
| Colony A | growth | no growth | ||
| Colony B | growth | growth | ||
| Colony C | no growth | no growth | ||
| Colony D | no growth | growth | ||
| Your colonies (1, 2, 3, etc.) |
Make sure you understand the phenotype and genotype of the controls first (ask a demonstrator if you help to understand). Then work through colonies A-D – record the genotype and predict the gene editing outcome of these colonies. These represent the four possible phenotypes that could arise from our experiment and doing this first should help when analysing the phenotype of your colonies.
Familiarise yourself with the growth of the controls first
A good place to start when analysing the results of a screen is to observe the growth of the control colonies and check that their phenotype is as expected. This is what controls are for – to prove that your experiment was technically successful and that the observed results were not a result of human or experimental error. If your controls do not perform as expected, you should still attempt to analyse the screen as best you can but be aware that it may not be possible to draw conclusions from your data.
Analyse your colonies
Methionine auxotrophy screen. The analysis of methionine auxotrophy is not as straightforward as simply checking whether there is growth or no growth. Yeast cells will already contain small amounts of methionine which will allow them to grow for a short period of time (until the methionine runs out). In addition, dead/dying yeast cells release methionine which can be taken up by surviving cells. This means that, at high densities, a growth phenotype can be hard to see. This is why we make serial dilutions of the colonies and compare the serial dilutions.
An example of met auxotrophy screening of positive and negative controls can be seen in the figure below.
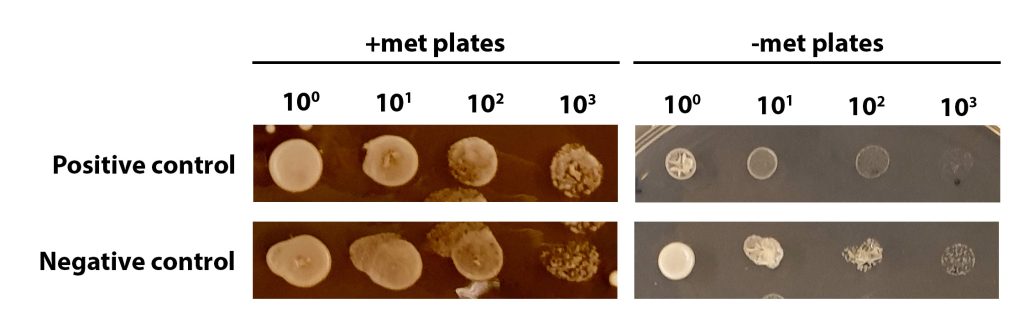
When analysing your plates remember:
- The negative control is the parental yeast strain. The genotype is NAT– Met+
- The positive control is a successfully edited yeast strain. The genotype is NAT+ met–
On the +met plates, you can see growth at all dilutions, with the density of cells lessening at higher dilutions. The growth at each dilution is similar for both positive and negative control yeast.
On the –met plates, the negative control shows growth at all dilutions (a similar pattern of growth to that seen on the +met plates). This is because the negative control (the parental yeast strain) has a Met+ phenotype, and should be able to grow on –met plates. The positive control is a successfully edited yeast cell and so is met– (unable to grow without methionine). You can see some growth of undiluted yeast cells (100) but serial dilutions result in little/no growth, particularly when compared to the negative control.
If you are unsure, talk to a demonstrator but it is OK to make a judgement call and it doesn’t matter if you get it wrong! Analysing the phenotype is just the first step in an experiment like this and we would confirm the genotype using molecular approaches, such as PCR. We will do an activity today looking at this.
Nourseothricin resistance screen. Observe the plates from the last session and note whether each colony is resistant to nourseothricin (NAT+) or sensitive to nourseothricin (NAT−). This should be more straightforward to analyse than the methionine auxotrophy screen − if the colony grows it is resistant; if it does not grow, it is sensitive.
Add the growth phenotype from your colonies to the table; fill in the genotype of the colonies that you screened; and predict the gene editing outcome.
- Do you have any colonies that have been successfully edited? What evidence do you have to support this?
- For colonies that have not been successfully edited, what is your best guess at the genotype and how do you think this arose?
PCR Primer Design Activity
As alluded to above, phenotype screening would be followed up by molecular screening that would confirm (or not!) the genotype predicted from the phenotype. A common way of screening for potential gene editing events is Polymerase Chain Reaction (PCR). You learnt about PCR at Level 1, so I don’t want to include a lot of detail here. If you would like a reminder, you can try this video by Khan academy (10 minutes) or you can read the same information in written form.
You will be given a printout to work through during the session (it will also be available on [the VLE] in case you miss the session / lose the printout). [see Resources for Teachers section]
